

![]() Pathogen Genomics Center, National Institute of Infectious Diseases.
Pathogen Genomics Center, National Institute of Infectious Diseases.
Analysis of whole genome sequences provides information that is useful for tracing back infection using genome-wide SNVs among clusters. During the primary wave, local public health centers were able to identify likely COVID-19 patients and their close contacts within location-specific clusters by conducting active epidemiological surveillance using the keywords “Wuhan, Hubei, and China.” Just as the situation had begun to improve around mid-March, a large number of new COVID-19 patients were diagnosed, many of with unclear infection routes. Tracing the infection routes was difficult because some Japanese cases had no recent history of travel to China or any country outside Japan. The SARS-CoV-2 haplotype network analysis suggested that the second wave of COVID-19 cases could have been imported via the returnees from EU, North America, or other countries.
This report suggests that haplotype network analysis using genome-wide SNVs can support epidemiological field investigations and highlight potential infection linkages. A project to determine viral evolution and its spread across the community is being conducted by researchers in the US and UK 10,11. To mitigate the next wave of COVID-19 in Japan, it is necessary to formulate a more efficient containment strategy.
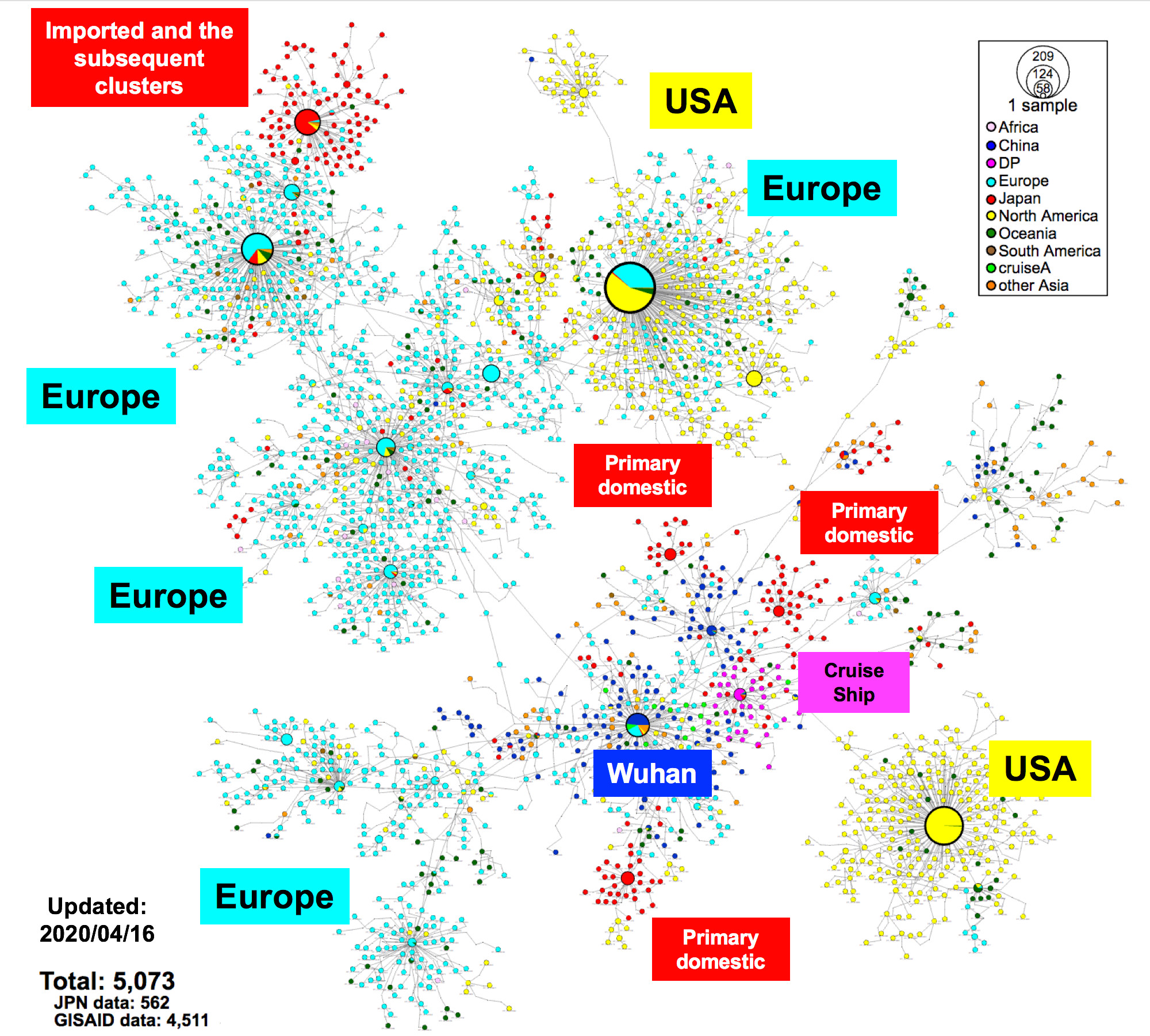
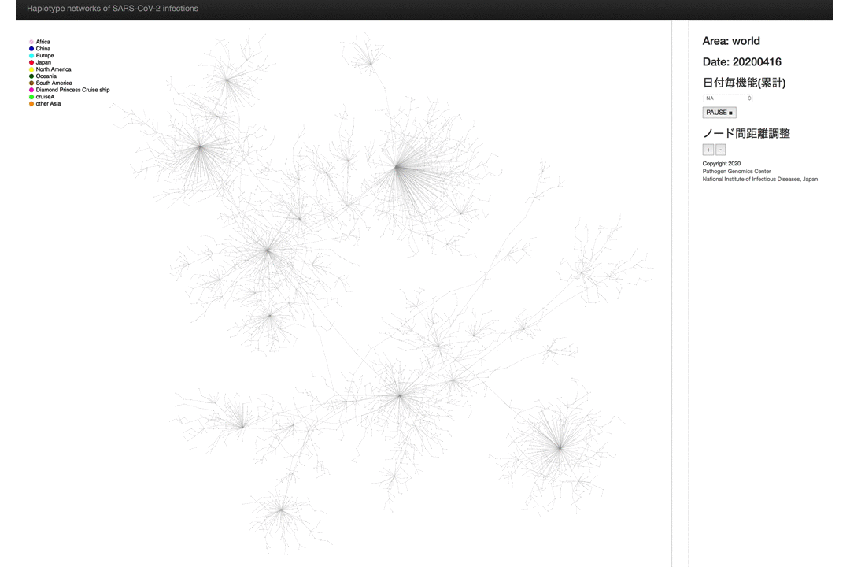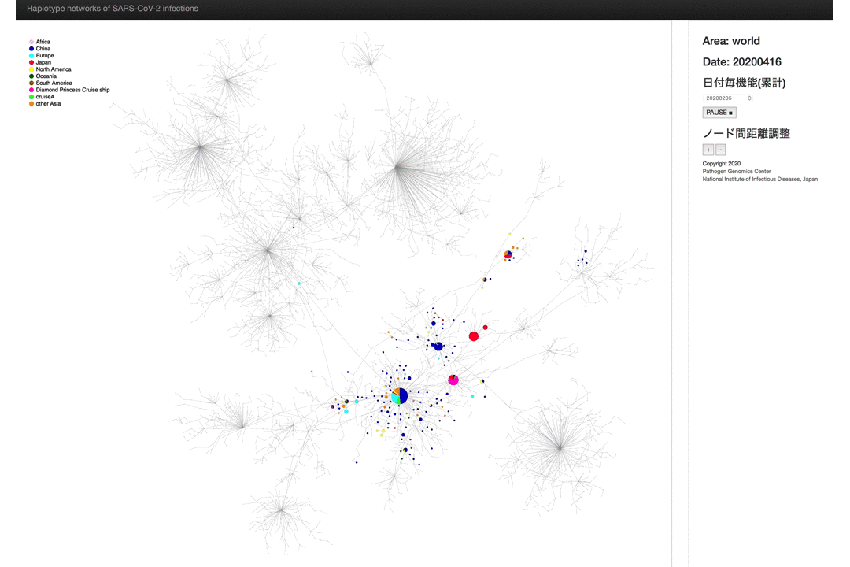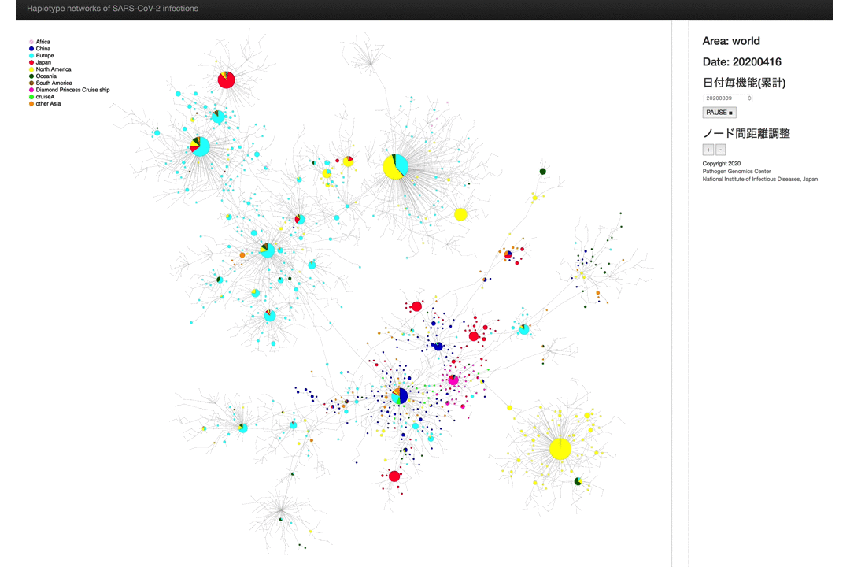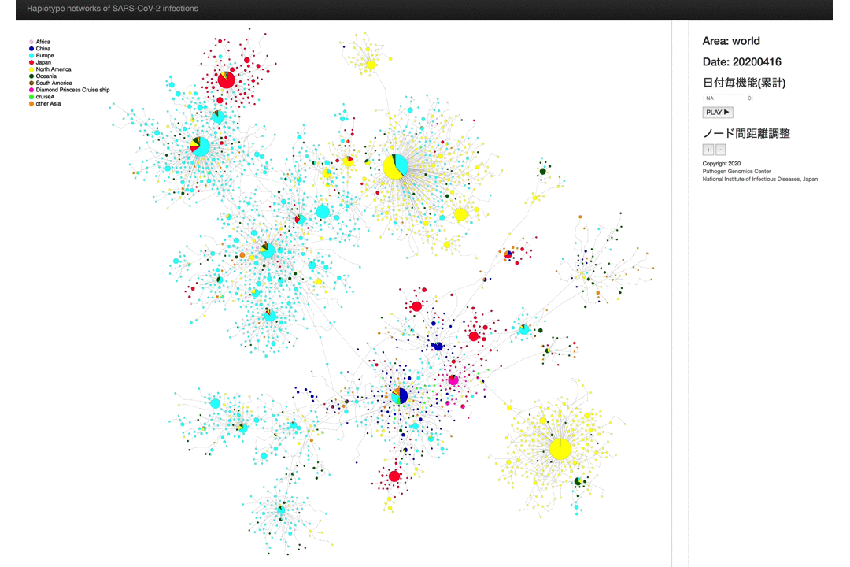
Fig. 1. Haplotype network analysis using genome-wide single-nucleotide variations (HN-GSNVs) of worldwide SARS-CoV-2 isolates.
Whole-genome sequences of SARS-CoV-2 isolates in Japan (n = 562) were compared with all GISAID-available SARS-CoV-2 genomes (n = 4,511, updated on April 16, 2020) using median-joining SNV network analysis. SARS-CoV-2 that disseminated from Wuhan City, China, at the end of December, 2019 (one of the potential origins of Wuhan-Hu-1) is plotted at the center of the haplotype network.
* An animated-gif file that allow you to observe the above network in chronological order.
{kind=link}
Acknowledgements:
We sincerely thank the COVID-19 working group in Japan listed below for the collection and transportation of clinical specimens. We are particularly grateful to the staff of the National Institute of Infectious Diseases, Field Epidemiology Training Program (FETP) team, the Ministry of Health, Labor and Welfare, and local governments for their assistance with administrative matters, field investigations, data collection, and laboratory testing. We would like to thank all the researchers who have kindly deposited and shared genomic data on GISAID. This study was supported by a Grant-in Aid from the Japan Agency for Medical Research and Development (AMED) under Grant Numbers JP19fk0108104 and JP20fk0108103).