国内で分離された腸管出血性大腸菌の全ゲノム配列データベース化と食中毒事例の解析
(IASR Vol. 42 p97-98: 2021年5月号)
腸管出血性大腸菌(enterohemorrhagic Escherichia coli:EHEC)の国内サーベイランスでは, 反復配列多型解析(MLVA)法やパルスフィールドゲル電気泳動(PFGE)法といった分子型別手法を用いた菌株間の比較が行われている。近年, 高速シークエンサーの実用化により, 集団感染等の調査に全ゲノム配列を用いた解析が利用可能となっている。EnteroBase(http://enterobase.warwick.ac.uk/)等の公共データベースでは, 15万件以上の大腸菌ゲノム配列が登録され, 国内外の株との比較解析が可能である。そこで, 公共データベースや自製データベースに存在する大腸菌ゲノム配列との比較を容易にするために, core genome single nucleotide polymorphism(cgSNP)およびcore genome multilocus sequence typing(cgMLST)解析環境の構築と, 国内EHECゲノムのデータベース化を行った。さらに同環境を利用し, 2020年に発生した食中毒事例について解析を行った。
cgSNPおよびcgMLST解析環境の構築とデータベース化
Core genomeとは, 対象となる菌株セットにおいてすべての菌株が有するゲノム領域を指す(99%を超える株が持つ領域をhard core, 95-99%の株が持つ領域をsoft coreとする定義も存在する)。これらの領域に存在する単一塩基多型(SNP)を抽出したものがcgSNPであり, 高精度な系統解析や菌株間の遺伝的距離の解析に用いられている。cgSNP抽出には, BactSNP等のソフトウェアを利用して行うが, 解析菌株を追加・削除した際の再解析に時間を要するなどの課題があった。そこで, 当部ではBactSNPおよびSnippyを組み合わせ, 迅速に再解析が可能な解析パイプラインを構築し, 当部でゲノム解読済みの約3,000株のEHECについてデータベース化を行った。一方cgMLST解析は, 数カ所のハウスキーピング遺伝子の配列多型解析であるMLSTを全ゲノムレベルに拡張したものである。大腸菌では2,513遺伝子を対象とする手法がEnteroBase上で公開されている。cgMLSTによって算出される菌株間の距離はcgSNPと高い相関を有し, かつ計算負荷が低いため, 多数の株から近縁な株を迅速に見つける目的に適している。現状では, cgMLST解析をするにはEnteroBase等の公共データベースへの登録か商用ソフト(BioNumerics)の利用が必要である。当部では, 集団感染株などが得られた際に公共データベース内の株から近縁な株を迅速に抽出するために, ローカルコンピュータで利用可能なcgMLST環境を構築し, 上記cgSNPと同様に国内EHECのデータベース化を行った。
2020年食中毒関連株の解析
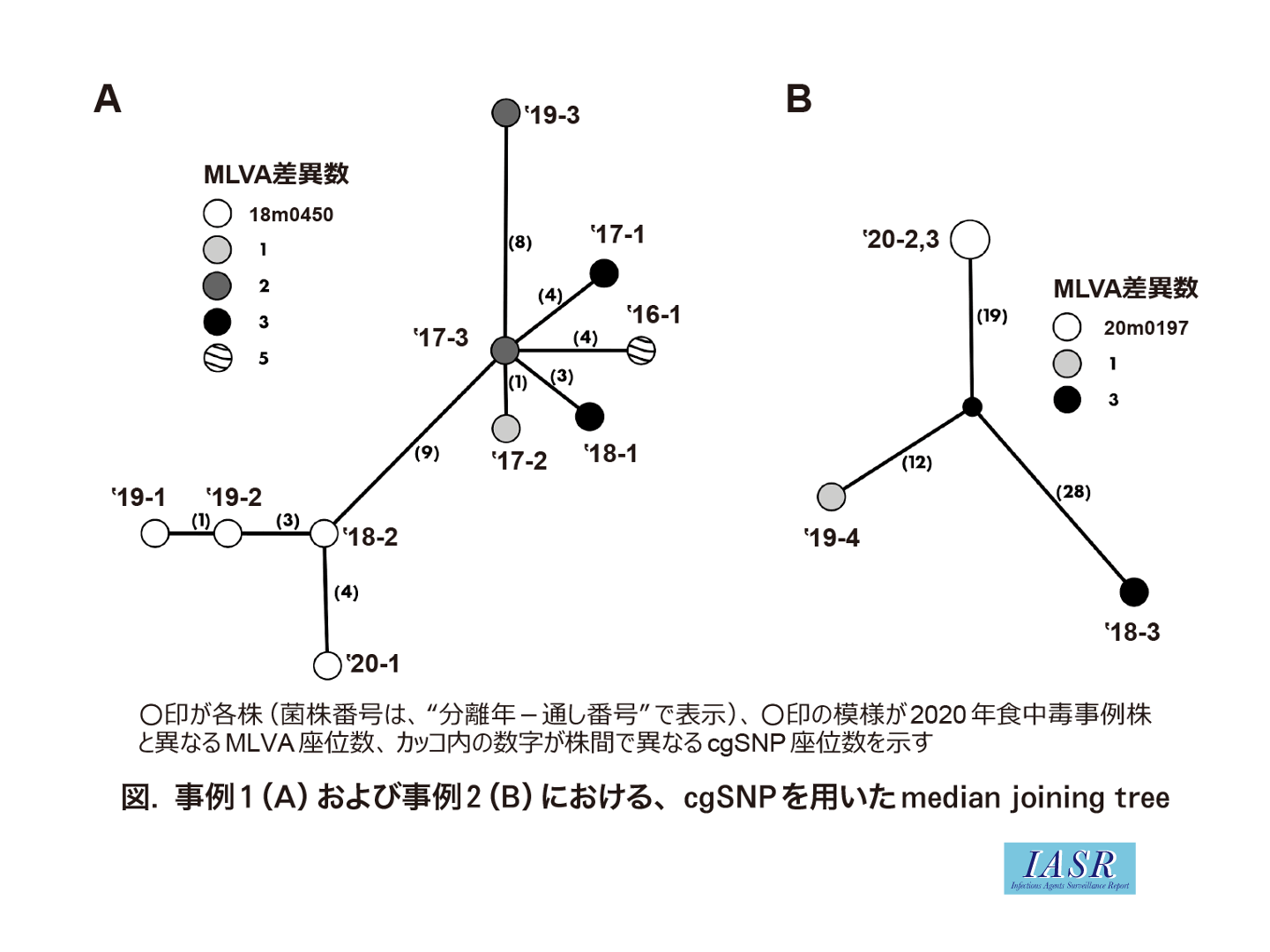
上記環境を用いて, 2020年に発生した2事例について代表株のゲノム解析を行い, 国内外のデータベース株との比較を行った。まず, 当該株のcgMLSTを行い, 近縁な株(30座位以内の差異)を抽出してcgSNP解析を行った。事例1では, cgMLSTにおいて食中毒関連株(’20-1, MLVA型:18m0450)との差異が30座位以内の株は, 国外株を含む44株であった。このうち, cgSNPの差異が20カ所以内だった株はMLVAでも近縁であった国内由来9株のみであった(図-A)。MLVA型が同一であった3株は最大で8カ所のcgSNPのみ認められ, 関連性が非常に高いことが示された。事例2では, cgMLSTにおいて食中毒関連株2株(’20-2および’20-3, MLVA型:20m0197)と30座位以内の差異が認められたのは国内株のみであった。これらの株についてcgSNPでの再解析を行ったところ, MLVAで1または3座位異なる2株とは30カ所以上のcgSNPが認められ, ゲノム既解読株の中から近縁な株は見出されなかった(図-B)。
{kind=link}
考 察
現在大腸菌では15万件以上の全ゲノム配列情報が公共データベースに蓄積されている。データベースに存在する情報は, 分離地や年代が偏っている可能性があり注意が必要であるものの, 有効に活用することによって国内外のEHECの伝播経路をより詳細に明らかにできると考えられる。本稿で取り上げた事例1では, 比較的近縁な株が数年前から継続的に分離されていることが示唆された。一方事例2のような事例では, 散発事例株や食品・動物由来株のゲノム解析によって, 菌株の由来が明らかになる可能性がある。また, EHEC感染源の特定には, 疫学情報が欠かせない。より正確なEHECの伝播経路解明のためには, MLVAやゲノム情報等の分子疫学情報と疫学調査のさらなる連携が求められる。