梅毒トレポネーマの分子型別: その意義と成果
(IASR Vol. 44 p190-191: 2023年12月号)目的と意義
梅毒は有効な治療方法が確立しているにもかかわらず, 世界的な流行を繰り返し, 2012年頃から拡大中の再流行には現在歯止めがかかっていない。このことは, 梅毒の制御には個々の症例の治療以上に, サーベイランスを通じた感染ルートの科学的根拠を持った推定, それによるリスク集団の特定と, それへの介入が重要であることを示している。これ以上の細分化はできないという意味での究極形としてのゲノム解析を含む分子型別は, その結果を元に感染ルート推定, リスク集団特定を行ううえで必須の段階であり, 推定結果に客観的根拠を付与する点で重要な意義を持つ。これを実効的に遂行するには, この解析による病原体のプロファイルデータの集積を継続的に行っておくことも重要である。
しかるに, 梅毒起因菌である梅毒トレポネーマ(Treponema pallidum: T. pallidum)は基本的に試験管内培養不能のため病原体の解析が困難で, 他の病原体で一般化しつつある分子型別およびゲノム解析データ集積が立ち遅れている。
分子型別を行ううえで不可欠な病原体の検出に関し, 早期診断を第一義として, 従前の抗体検査に加え, 抗体陽転までのタイムラグが無く, 直接病原体DNA検出が可能なPCR等での確定診断がある程度普及してきた。
これを背景に, 病原体DNA陽性の検体を流用し, 試験管内培養不能の当該菌でも, いくつかの多型性遺伝子をターゲットにしたPCR産物解析による分子型別が2010年代から本格的に開始された。この型別は「感染ルートの科学的根拠をともなった推定」へつながることが期待されていた。実際の手法として, 実施数での実績が有るものとしてECDC(Enhanced CDC)typing1), SBMT(Sequence Based Molecular Typing)2), MLST(Multi-Locus Sequence Typing)3)等が挙げられる。これらは, 一定の分解能を備え, また参照株との比較により系統分類能もある程度備えるとされるが, いくつかの特定遺伝子の情報のみで判断するため, ある頻度で起こる組み替えや突然点変異等を考慮した場合, 誤った系統分類をする可能性は否定できない。
試験管内培養不能のT. pallidumにおいて全ゲノム解析は立ち遅れてきたが, 参照株全ゲノム情報を持つターゲットキャプチャー分子を用いた選択濃縮法により, 高コスト, 低成功率ながら, この解析も検体から実行可能になってきた。これにより菌株の詳細プロファイルとその株間差情報の獲得が期待でき, 「科学的感染ルート推定」の現実性が増している。
これまでの経験と実績
我々は2012年より病変部からのT. pallidum DNA検出と病原体の分子型別・ゲノム解析に着手し, いくつかの知見を得て報告している。
まず, 我々と連携したクリニックからの疑い検体が急増した2017年の国内検体群についてECDC typing解析を行い, 結果を報告した4)。この解析では, 国内のheterosexual患者由来検体の分子型は非常に均質性が高く, これに対してMSM(men who have sex with men)由来のそれはかなり多様な分子型に分散していることがわかった。
続いて, 2014~2018年の国内検体を対象に全ゲノム解析を試行し, データ取得ができた20例につき, そのデータを元に系統樹を作成5), 前述のECDC typing結果を踏襲し, heterosexual患者由来の分子型の均質性とMSM由来のそれの多様な分子型への分散を再確認した5)。また, データバンクで公開されていた各国の検体のデータと比較し, 国内heterosexual患者由来検体が外国株では中国株と最も近縁であることが判明した。しかし, Bayesian maximum credibility phylogenetic analysis6)を用いた計算では, 両国株群の仮想共通祖先株の存在時期は2007年と算出され, 一般に議論されているよりもかなり以前に両国株が分岐完了していると推定された5)。
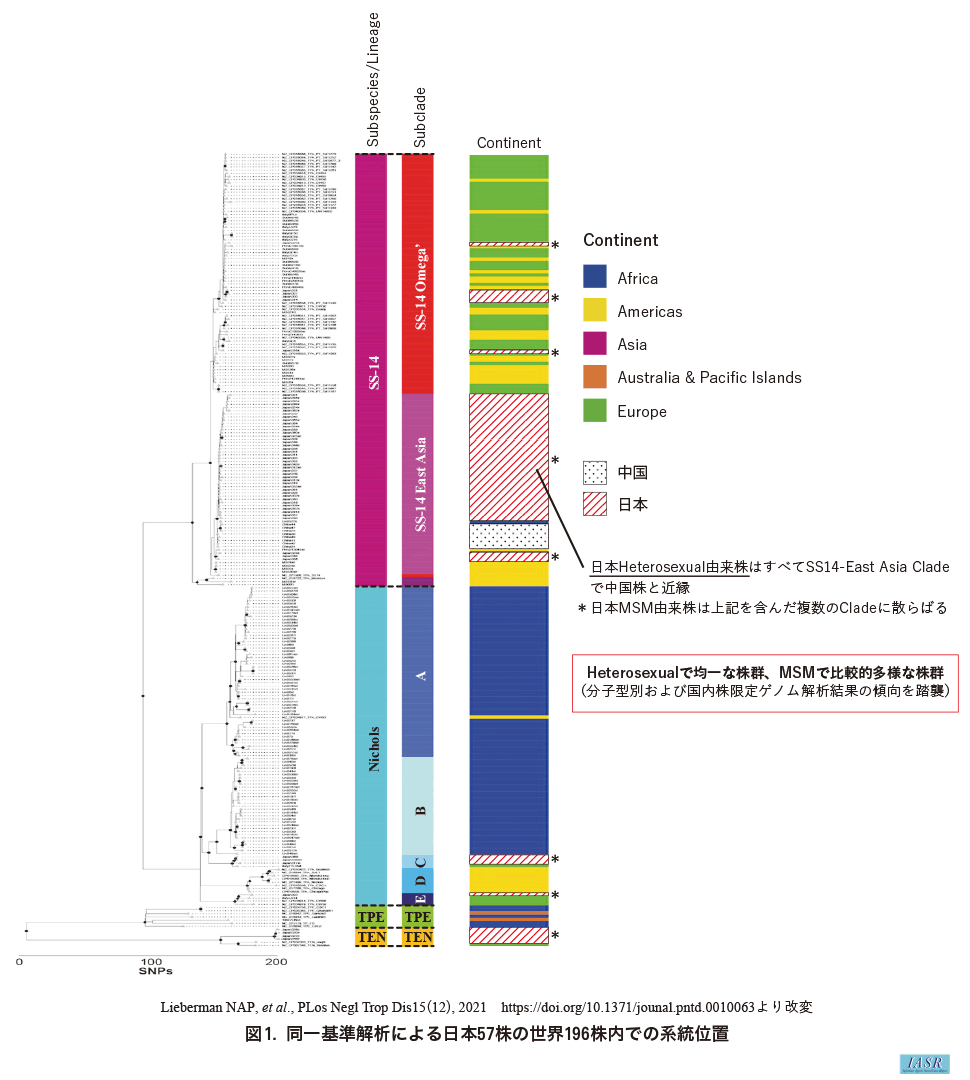
さらに, 2019~2020年の国内検体57例を含む世界株196例の統一基準解析を行い, 前者が後者全体の中で占める系統位置を把握した7)(図1: webのみ掲載)。この解析でも, 国内のheterosexual患者由来検体の系統の均質性と中国株との近縁性, およびMSM由来のそれの系統の比較的な多様性, が再確認されている。
{kind=link}
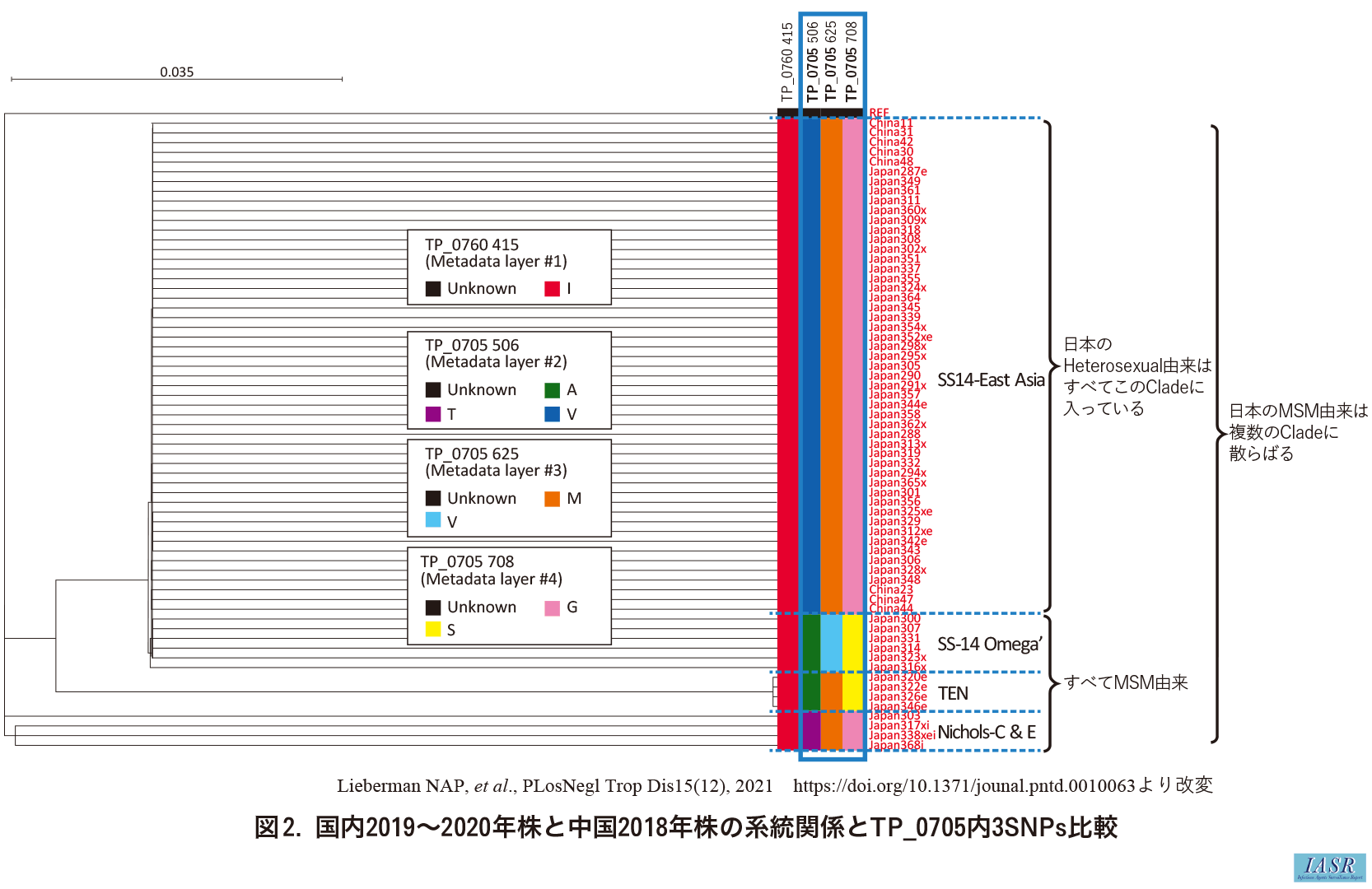
また, この解析での国内株のみに着目し, 上記報告7)での系統分類に従うと, 大きく4系統「SS14-East Asia」, 「SS14-Omega'」, 「TEN」, 「Nichols-C&E」(細かくは5系統だが, 「Nichols-C」と「Nichols-E」の分割は多様なNichols系統株の便宜的細分の意味合いが強い7))に分類され, heterosexual患者由来はすべて「SS14-East Asia」に属することが判明した。また各系統とそのゲノムデータとを比較し, 単一遺伝子TP_0705内の3カ所のSNPs(Single Nucleotide Polymorphisms)組み合わせが系統と一意対応することが判明した(図2: webのみ掲載)。これは, ゲノム解析不成立でも, この遺伝子の解析で国内株系統を少なくとも推定できる可能性を示し, 簡便系統分類法の観点で有望である。現在, 2021年以降の株でも, この対応関係が維持されているか検討を継続している。
{kind=link}
参考文献
- Marra C, et al., J Infect Dis 202: 1380-1388, 2010
- Vaulet LG, et al., PLoS ONE, 2017
https://doi.org/10.1371/journal.pone.0172905 - Pospíšilová P, et al., PLoS ONE, 2018
https://doi.org/10.1371/journal.pone.0201068 - Kanai M, et al., J Clin Microbiol 57, 2019
https://doi.org/10.1128/jcm.01167-18 - Nishiki S, et al., Sci Rep, 2021
https://doi.org/10.1038/s41598-021-82337-7 - Bouckaert R, et al., PLoS Comput Biol 15, 2019
https://doi.org/10.1371/journal.pcbi.1006650 - Lieberman NAP, et al., PLos Negl Trop Dis 15, 2021
https://doi.org/10.1371/journal.pntd.0010063